
Introduction:
QSAR techniques are contributing in a major way to drug design by improving the potency, efficacy, and selectivity of lead compounds in drug design. This is also true for drug design for diabetes, where techniques like virtual screening and 3D-QSAR are helping scientists design drugs with reduced side effects and increased efficacy and potency.
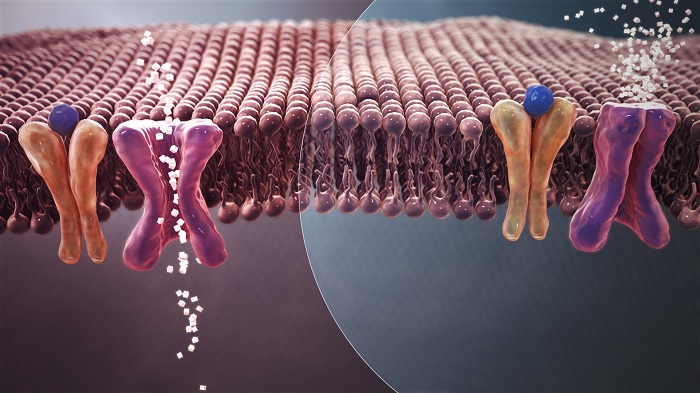
Figure 1: Mechanism of normal Blood Sugar (white crystals) absorption (Left) Vs. insulin resistance in Type 2 Diabetes (Right).
According to an estimate by the ‘International Diabetes Federation’, presently about 9.3 per cent of the world’s population in the age group of 20-79 years suffers from diabetes, which is estimated to increase to 10.9 per cent by 2045[1], [2]. Inflammation, epigenetics, and autophagy are considered the three main factors responsible for the onset of diabetes.[3], [4]The main questions that antidiabetic drug discovery faces today are: how to encounter insulin resistance, alternatives to the parenteral route of administration as patient compliance for it is an issue, low bioavailability, and immediate release of oral drugs for diabetes due to which dosage frequency has to be increased, designing drugs that do not show many adverse effects and can target metabolic syndrome-X along with diabetes.[5]
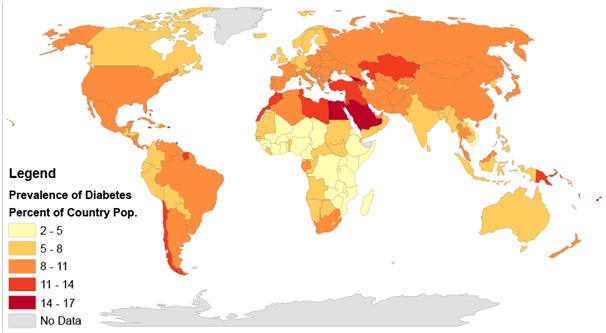
Figure 2:Rates of diabetes worldwide in 2014. The worldwide prevalence was 9.2 per cent. In India, it is 8.9 per cent as of 2020.
(https://en.wikipedia.org/wiki/Diabetes_in_India#/media/File:Prevalence_of_Diabetes_by_Percent_of_Country_Population_(2014)_Gradient_Map.png)
QSAR and drug targets for Diabetes:
The multidirectional and translational nature of antidiabetic drug discovery makes it ideal for QSAR studies. Evidence does point out that QSAR approaches would be a chief player in antidiabetic drug discovery. Predominantly, QSAR investigations have been carried out on five chief drug targets in diabetes, viz., peroxisome proliferator-activated receptor (PPAR), sodium-dependent glucose cotransporter 2 (SGLT2), protein tyrosine phosphatase 1B (PTP-1B), dipeptidyl peptidase IV (DPP-IV) and glycogen synthase kinase 3β (GSK-3β). Various QSAR models have been designed for PPAR full agonists to develop insulin-sensitising PPAR modulators that have very few adverse effects. Many QSAR studies have been performed on another attractive drug target for diabetes, PTP-1B, however, its high homology with T-cell protein-tyrosine phosphatase (TCPTP) complicates the drug design. For the design of the inhibitors of DPP-IV, GSK-3β, and SGLT2, mainly comparative molecular field analysis (CoMFA) and comparative molecular similarity analysis (CoMSIA) have been used in 3-D QSAR models.
In a conventional QSAR method, depicting the 3-D structure or stereochemistry of molecules is difficult. In addition to that, a classical QSAR model has insufficient parameters to describe drug-receptor interactions. Due to this, most of the antidiabetic drug discovery studies focus on 3D-QSAR approaches, which associate macroscopic target properties with computed atom-based descriptors built on the three-dimensional structure of the molecule. Due to this, the lead optimisation becomes quite efficient. This can be applied to both on-target activities and off-target activities. Classic ligand-based methods like CoMFA and CoMSIA lag behind QSAR models that are based on receptors like COMBINE, AFMoC, and CoRIA. In addition to that, one has to be careful while applying various QSAR methods to circumvent misinterpretation, particularly for conformational sensitivity and alignment dependency [6].
How QSAR is helping design novel anti-diabetic drugs?
Managing the off-target effects of hypoglycemic agents is an important challenge for anti-diabetic drug discovery. Here, QSAR methodologies have helped scientists to understand the association between molecular properties and hypoglycemic activity of the drug molecule. But the insights from QSAR must be combined with other computational techniques to design novel anti-diabetic scaffolds, which are highly targeting selective and potent.
Diabetes is a complex disease affecting many organs and there are many drug targets. Defining minute differences at the molecular level that affect activities such as target bioactivity, solubility and toxicity is another challenge. In QSAR, the trend for anti-diabetic drug discovery is to focus on a single structural feature of the compound and explore the differences among a fixed data set. Then the improvement in activity is sought by making use of features like electrostatic, steric, and hydrogen bonding. But this does not work in mixture derivatives. Moreover, the quality of a QSAR method is directly proportional to the quality of the experimental data. The beauty is that a good quality QSAR model can eventually lead to the development of new ligands that have not been obtained synthetically or no experimental testing has been done on them. In addition to that, the experimental data should be sufficiently homogeneous for good predictive power and for enriching the affinity database of the inhibitors. Another aspect that needs to be taken care of is ‘validating the QSAR models’ which will include a selection of training set compounds, setting training set size and impact of variable selection on the quality of prediction [7].
It is to be understood that the basic knowledge of ligand-protein interactions among various drug targets and ligands in diabetes is not very sophisticated. There are many evidence gaps in there, such as a lack of high-resolution structural knowledge that hampers the predictive power of a QSAR model designed for anti-diabetic drug discovery. This is because the precision and accuracy will rely on the number of structures serving as a template and their homology to the protein of interest. In the case of anti-diabetic drug discovery, the experimental 3-D structures of most of the inhibitors are unidentified. So careful consideration of such studies is needed.
The other issue that needs consideration is the selectivity and diversity of ligands in anti-diabetic drug discovery. For instance, protein tyrosine phosphatase is a drug target for diabetes and there are a variety of ligands that can bind to it in diverse ways. A 3D-QSAR model thus will have to predict new binding pocket structures for a diverse and selective set of ligands. So finally, some rules of thumb in QSAR that should be followed for designing novel inhibitors for diabetes drug targets are large and informative datasets, clarity and simplicity in the methods correlating activity and structural descriptors, and effective evaluation of the models.
Even if structural models are available to describe ligand-receptor interactions, because of the dynamic nature of the ligand recognition process, it is problematic to develop a good 3-D QSAR model. In addition to that, the combination of QSAR models with other molecular modeling techniques shows more promise for anti-diabetic drug design, as to date there is no evidence of any antidiabetic agent designed solely by QSAR methods. The validation of the QSAR models by experiment, i.e. by developing novel bioactive molecules, is missing in most of the studies [8].
Conclusions:
The development of multiple QSAR models that simultaneously work on multiple drug targets for antidiabetic drug discovery can be a game-changer. In addition to that, ADME-curated training lists for QSAR models should be accommodating. There is a lot of communication gap between academia and industry and much needs to be done for the translation of findings from QSAR models to marketable drugs. In addition to that, we need to stress the 'in Cerebro-in silico’ approach, i.e. the method applied should be specific to the target-ligand system.
Visit now: https://www.pharmafocusasia.com/articles/qsar-changing-the-landscape-of-antidiabetic-drug-discovery




















Comments
0 comment