

One significant application of long-read sequencing technology is the optimization of genome assembly processes. The current sequencing technologies have achieved read lengths sufficient to cover and span the majority of repetitive structures within genomes, providing robust support for accurate genome assembly. However, a prevailing challenge arises when dealing with diploid genomes, such as the human genome. This challenge involves achieving accurate haplotype resolution from telomere to telomere without the guidance of a reference genome, thereby ensuring the completeness and accuracy of the genome assembly.
De Novo Genome Assembly
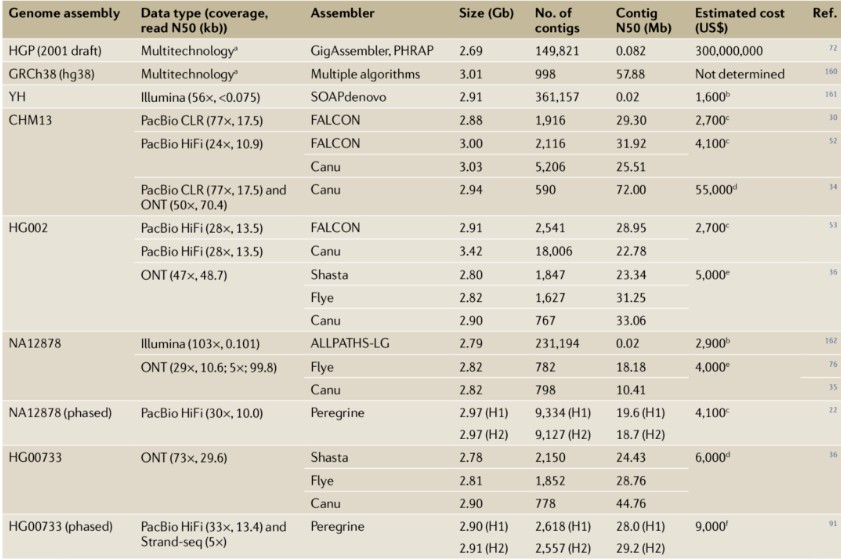
De novo genome assembly aims to accurately determine the sequence of each base within a genome by reassembling randomly sampled sequence fragments. These concatenated sequence fragments, based on their overlapping regions, are referred to as contigs. Ideally, each chromosome should be represented by a single contig. However, short-read sequencing technology has faced challenges in assembling mammalian genomes due to the difficulty of bridging repetitive sequences, often resulting in numerous gaps. Extensive research has demonstrated that the continuity of genome assemblies derived from long-read sequencing significantly surpasses that of traditional short-read sequencing and Sanger sequencing methods (Table 1).
For instance, in early 2015, the average contig N50 of 99 mammalian genome assemblies in GenBank was merely 41 kb, none of which utilized long-read sequencing as the primary data type. By early 2020, GenBank included over 800 genome datasets employing PacBio or Oxford Nanopore Technologies (ONT) data, with contig N50 lengths exceeding 5 Mb. These datasets encompass several early human genome projects, such as NA12878, CHM13, HX1, and AK1. The substantial improvement in assembly continuity is attributed not only to the increased read lengths but also to the development of software specifically designed for long-read sequencing data (e.g., Canu, HiCanu, Peregrine, FALCON, Flye, wtdbg2, and Shasta) and the application of auxiliary tools like optical mapping (e.g., Bionano Genomics) and electronic mapping (e.g., Nabsys).
It is noteworthy that individual laboratories can now achieve human genome sequencing and assembly with continuity levels comparable to or exceeding those of the Human Genome Project within a few weeks (Figure 1a). For example, Shafin et al. constructed 11 highly contiguous human genome assemblies (median NG50 of 18.5 Mb) using data from three PromethION chips and six hours of computation time on a machine equipped with 28 CPU cores and over 1 TB of RAM. Similarly, Chin and Khalak completed a human genome assembly in less than 100 minutes (equivalent to 30 CPU hours, excluding initial computation costs for generating PacBio HiFi reads) with an N50 contig length exceeding 20 Mb, relying solely on PacBio HiFi data. In contrast, aligning Illumina short-read human genome sequencing data, with approximately 30x sequencing depth, typically requires over 100 CPU hours.
Table 1: Case Studies of Human Genome Assembly Using Long-Read Sequencing
Genome Assembly Polishing and Phasing
The polishing and phasing steps are critical in the genome assembly process. Despite the pursuit of speed, long-read genome assemblies must undergo rigorous correction and evaluation to ensure performance comparable to that of genomes generated through Illumina or Sanger sequencing. Unpolished genome assemblies tend to contain numerous small insertion-deletion errors, complicating genome annotation significantly. To address these issues, various polishing tools have been developed, including Racon, Nanopolish, MarginPolish, HELEN, Quiver, Arrow, and Medaka. These tools utilize short-read sequence data from the same individual for error correction, effectively resolving most such issues.
Advancements in algorithms and the emergence of high-accuracy long-read sequence data types, such as HiFi data and Nanopore Q20 chemistry, have reduced the dependency on short-read data for polishing to a certain extent. Currently, a primary development direction is the generation of high-quality, fully-phased diploid genomes, where both haplotypes represent portions of the human genome. Essentially, haplotype-resolved genomes transform the 3 Gb human genome into a 6 Gb genome, containing complementary information from both maternal and paternal sources, thereby significantly enhancing the overall sensitivity of variant detection.
Fortunately, phased de novo genome assembly has now become feasible, owing to the application of novel strategies such as trio binning, which utilizes parental information for phasing long reads. Concurrently, computational methods leverage the inherent phasing properties of long-read data (e.g., FALCON-Unzip) and apply orthogonal techniques to phase single nucleotide polymorphisms within long-read data (e.g., Strand-seq, Hi-C, and 10x Genomics). The fundamental concept involves physically or genetically phasing a single genome, thereby partitioning the long-read data into two distinct parental genome datasets for independent assembly. This approach is particularly crucial for resolving structural variants and their haplotype structures, as structural differences between haplotypes often result in assembly graph heterozygosity or unassembled regions, which fail to accurately reflect the sequence and are thus biologically insignificant.
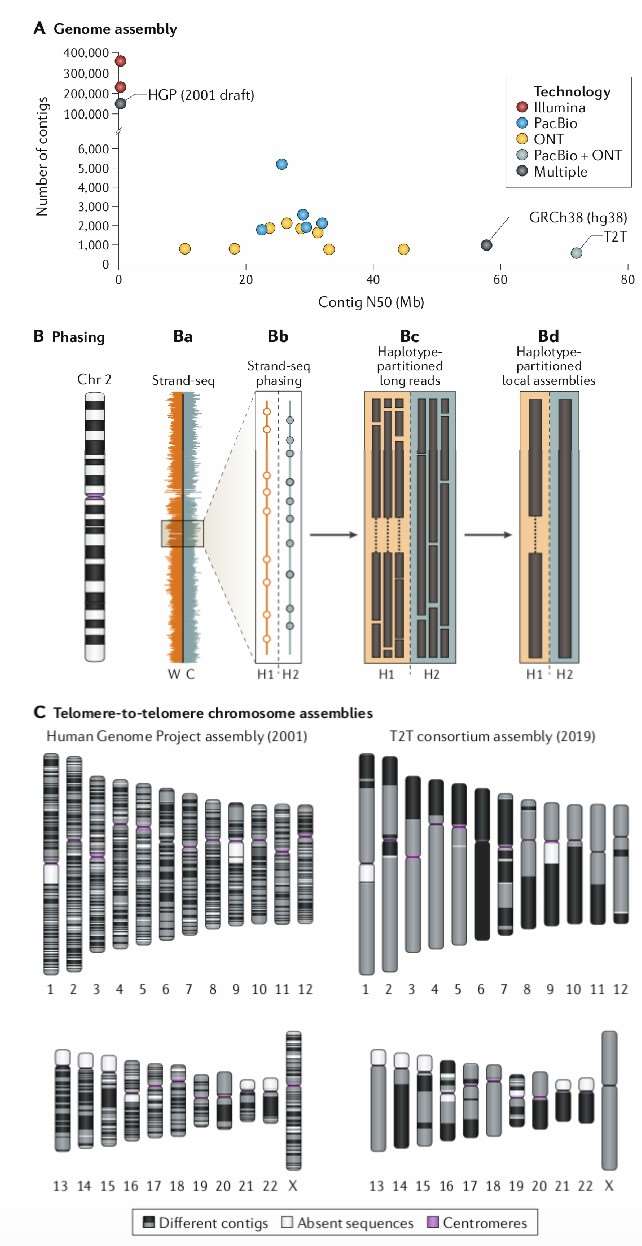 Figure 1: Enhancing Human Genome Assembly with Long-Read Sequencing
Figure 1: Enhancing Human Genome Assembly with Long-Read Sequencing
Telomere-to-Telomere Chromosome Assemblies
The ultimate objective of genome assembly is the construction of complete contigs for each chromosome, ensuring an accurate sequence from telomere to telomere without any gaps. Currently, more than half of the remaining gaps in genome assemblies are attributable to segmental duplications, which can be readily identified by increasing read depth. These unassembled regions primarily result from highly similar sequences but can be effectively distinguished using paralogous sequence variant maps based on high-accuracy long-read sequences. Since its release in 2001, the human reference genome has been the gold standard for mammalian genomes and has received substantial investment over the past two decades to enhance its accuracy and continuity. However, even in the current iteration (GRCh38 or hg38), the number of contigs significantly exceeds the number of chromosomes (998 contigs corresponding to 24 chromosomes). Most of these gaps are associated with large repetitive sequences in centromeres, specifically pericentromeric DNA and segmental duplications.
The application of ONT (Oxford Nanopore Technologies) and PacBio (Pacific Biosciences) technologies to the nearly haploid CHM13 human genome indicates significant progress toward achieving telomere-to-telomere genome assemblies. Miga et al. successfully represented the CHM13 human genome with 590 contigs, including a complete telomere-to-telomere assembly of the X chromosome, by integrating these sequencing data types with advanced assembly algorithms. This achievement was primarily facilitated by generating high-coverage ultra-long ONT reads, which exhibit greater continuity than GRCh38 (81.3 Mb versus 57.9 Mb), and reconstructing the highly repetitive centromeric α-satellite array on the X chromosome for the first time. However, the telomere-to-telomere assembly process remains heavily reliant on manual intervention, with hundreds of collapsed repetitive sequences still requiring comprehensive genome-wide analysis. Nevertheless, current research efforts are focused on automating centromere assembly, utilizing tools such as CentroFlye and HiCanu.
Further development is necessary to routinely produce telomere-to-telomere chromosome assemblies, whether for processing and assembling PacBio HiFi sequence data or coupling it with ONT ultra-long reads using enhanced assembly tools. Achieving routine, accurate telomere-to-telomere assembly for diploid genomes may take several years, due to the high cost and extended generation time of specialized data types (i.e., ultra-long reads) and the inherent challenges posed by unexplored regions of the human genome. Many regions, including centromeres, acrocentric regions, and large segmental duplications, have not yet been correctly sequenced and assembled. Consequently, any computational assembly algorithms targeting these regions will require stringent validation and comprehensive evaluation.
References
- Logsdon et al., The structure, function and evolution of a complete human chromosome 8. 2021. Nature
- Miga et al., Telomere-to-telomere assembly of a complete human X chromosome. 2020. Nature
- Logsdon et al., Long-read human genome sequencing and its applications. 2020. Nat. Rev. Genet.



















Comments
0 comment